Difference between revisions of "ChemFoam"
(→Chemical reactions) |
(→Chemical reactions) |
||
| Line 443: | Line 443: | ||
} | } | ||
} | } | ||
| + | } | ||
| + | </cpp><br> | ||
| + | |||
| + | The remaining two quantities to define are the forward and backward reaction rate constants <math> k_f </math> and <math> k_r </math>. The forward rate constant is calculated for Arrhenius type reactions as follows (see also <ref> Poinsot, Thierry, and Denis Veynante. Theoretical and numerical combustion. RT Edwards, Inc., 2005.</ref): | ||
| + | |||
| + | <table width="70%"><tr><td> | ||
| + | <center><math> | ||
| + | k_f = A T^\beta exp(-T_a/T) | ||
| + | </math></center> | ||
| + | |||
| + | A is the per-exponent factor, Ta the activation temperature and beta is the exponent of the temperature in the calculation of the reaction rate. The soure code can be find in $FOAM_SRC/thermophysicalModels/specie/reaction/reactionRate/ArrheniusReactionRate/ArrheniusReactionRateI.H: | ||
| + | |||
| + | <br><cpp> | ||
| + | inline Foam::scalar Foam::ArrheniusReactionRate::operator() | ||
| + | ( | ||
| + | const scalar p, | ||
| + | const scalar T, | ||
| + | const scalarField& | ||
| + | ) const | ||
| + | { | ||
| + | scalar ak = A_; | ||
| + | |||
| + | if (mag(beta_) > VSMALL) | ||
| + | { | ||
| + | ak *= pow(T, beta_); | ||
| + | } | ||
| + | |||
| + | if (mag(Ta_) > VSMALL) | ||
| + | { | ||
| + | ak *= exp(-Ta_/T); | ||
| + | } | ||
| + | |||
| + | return ak; | ||
} | } | ||
</cpp><br> | </cpp><br> | ||
Revision as of 18:34, 23 February 2020
chemFoam
Solver for chemistry problems, designed for use on single cell cases to provide comparison against other chemistry solvers, that uses a single cell mesh, and fields created from the initial conditions.
1 Solution Strategy
This solver provides an excellent starting point for those who want to have a first impression on the influence of the chemical reactions in the equations describing the evolution of the species concentration and also the evolution of temperature (i.e. the energy equation). Since the computational domain consists only of one cell, the only mechanism influencing the evolution of the spices concentration and of the temperature are the chemical reactions. The system of equations which is solved is the following:
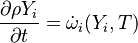 | (1) |
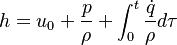 | (2) |
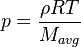 | (3) |
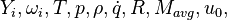 are the species mass mass fraction, the reaction rate, the temperature, the pressure, the density, the heat generated by the combustion, the gas constant, the average molar weight and the initial internal energy, respectively. Note that for a given enthalpy, pressure and density the temperature can be calculated.
are the species mass mass fraction, the reaction rate, the temperature, the pressure, the density, the heat generated by the combustion, the gas constant, the average molar weight and the initial internal energy, respectively. Note that for a given enthalpy, pressure and density the temperature can be calculated.
Like most of the solvers present in OpenFOAM also this solver follows a segregated solution strategy. That means that for each quantity of interest one linear equation is solved and the coupling between the equations is achieved by explicit source terms. Briefly summarized the solution is achieved as follows:
1) Solve the chemistry: The purpose is to get the reaction rates for each species involved and the heat realized by the chemical reaction
2) Solve the species equation: The purpose it to get the species concentration at the new time step
3) Solve the energy equation: Here we get the enthalpy (over the thermodynamics also the temperature) at the new time step
4) Solve the pressure equation: Required to calculate the enthalpy
The source code can be found in chemFoam.C
/*---------------------------------------------------------------------------*\ ========= | \\ / F ield | OpenFOAM: The Open Source CFD Toolbox \\ / O peration | \\ / A nd | Copyright (C) 2010-2011 OpenCFD Ltd. \\/ M anipulation | ------------------------------------------------------------------------------- | Copyright (C) 2011-2017 OpenFOAM Foundation ------------------------------------------------------------------------------- License This file is part of OpenFOAM. OpenFOAM is free software: you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation, either version 3 of the License, or (at your option) any later version. OpenFOAM is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details. You should have received a copy of the GNU General Public License along with OpenFOAM. If not, see <http://www.gnu.org/licenses/>. Application chemFoam Group grpCombustionSolvers Description Solver for chemistry problems, designed for use on single cell cases to provide comparison against other chemistry solvers, that uses a single cell mesh, and fields created from the initial conditions. \*---------------------------------------------------------------------------*/ #include "fvCFD.H" #include "psiReactionThermo.H" #include "BasicChemistryModel.H" #include "reactingMixture.H" #include "chemistrySolver.H" #include "OFstream.H" #include "thermoPhysicsTypes.H" #include "basicSpecieMixture.H" #include "hexCellFvMesh.H" // * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * // int main(int argc, char *argv[]) { argList::addNote ( "Solver for chemistry problems, designed for use on single cell cases" " to provide comparison against other chemistry solvers" ); argList::noParallel(); #define CREATE_MESH createSingleCellMesh.H #define NO_CONTROL #include "postProcess.H" #include "setRootCaseLists.H" #include "createTime.H" #include "createSingleCellMesh.H" #include "createFields.H" #include "createFieldRefs.H" #include "readInitialConditions.H" #include "createControls.H" // * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * // Info<< "\nStarting time loop\n" << endl; while (runTime.run()) { #include "readControls.H" #include "setDeltaT.H" ++runTime; Info<< "Time = " << runTime.timeName() << nl << endl; #include "solveChemistry.H" #include "YEqn.H" #include "hEqn.H" #include "pEqn.H" #include "output.H" runTime.printExecutionTime(Info); } Info << "Number of steps = " << runTime.timeIndex() << endl; Info << "End" << nl << endl; return 0; } // ************************************************************************* //
2 Chemical reactions
Elementary chemical reaction involving 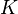 species in
species in 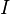 reactions can be expressed in the following form (see also https://www3.nd.edu/~powers/ame.60636/chemkin2000.pdf or the book of [1]):
reactions can be expressed in the following form (see also https://www3.nd.edu/~powers/ame.60636/chemkin2000.pdf or the book of [1]):
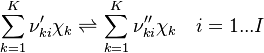 | (4) |
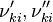 and
and 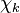 are the forward, the backward stoichiometric coefficient and the chemical symbol of the specie
are the forward, the backward stoichiometric coefficient and the chemical symbol of the specie 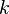 , respectively.
, respectively.
In order to make the notation more clear to those who start learning about the usage of CFD to simulate combustion process, Zel'dovich mechanism for the oxidation of N0 (see https://en.wikipedia.org/wiki/Zeldovich_mechanism or also the book of [2]) is used as an example:
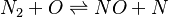 |
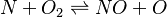 |
Based on the above example the forward 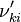 , the backward stoichiometric coefficient
, the backward stoichiometric coefficient 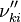 and the chemical symbol of the specie k can be written as follows:
and the chemical symbol of the specie k can be written as follows:
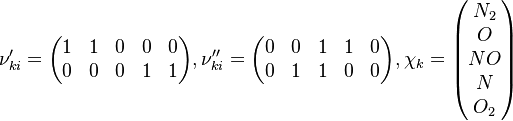 The reactions are specified in the file constant/thermophysicalProperties where the chemistry reader, the file containing the reactions and the thermodynamic properties have to be specified: /*--------------------------------*- C++ -*----------------------------------*\ | ========= | | | \\ / F ield | OpenFOAM: The Open Source CFD Toolbox | | \\ / O peration | Version: v1912 | | \\ / A nd | Website: www.openfoam.com | | \\/ M anipulation | | \*---------------------------------------------------------------------------*/ FoamFile { version 2.0; format ascii; class dictionary; location "constant"; object thermophysicalProperties; } // * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * * // thermoType { type hePsiThermo; mixture reactingMixture; transport sutherland; thermo janaf; energy sensibleEnthalpy; equationOfState perfectGas; specie specie; } chemistryReader foamChemistryReader; foamChemistryFile "<constant>/reactions"; foamChemistryThermoFile "<constant>/thermo"; //CHEMKINFile "<case>/chemkin/chem.inp"; //CHEMKINThermoFile "<case>/chemkin/therm.dat"; //CHEMKINTransportFile "<case>/chemkin/transportProperties"; // ************************************************************************* // For the above example the reactions look as follows: elements 2(O N); species 5(O O2 N2 N NO); reactions { un-named-reaction-0 { type reversibleArrheniusReaction; reaction "NO^1 + N^1 = N2^1 + O^1"; A 2.1e+10; beta 0; Ta 0; } un-named-reaction-1 { type reversibleArrheniusReaction; reaction "N^1 + O2^1 = NO^1 + O^1"; A 5.83e+06; beta 1.01; Ta 3120; } } In our example both reactions are reversible Arrhenius reactions. A is the per-exponent factor, Ta the activation temperature and beta is the exponent of the temperature in the calculation of the reaction rate (see below for further explanations). The values are taken from the book of [3]. Note that the values of the constants are calculated in the OpenFOAM native unit system of kmol, m3, s, K. Having established the chemical reactions which are desired to be solved, the next step is to describe the velocity at which the chemical reaction occur, i.e. the change of the concentration of the single species with time. The reaction rate
|
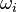 of the reaction i can be written as (see also the book
of the reaction i can be written as (see also the book ![\omega_i = k_{fi} \prod_k [X]_k^{\nu_{ki}'} - k_{ri} \prod_k [X]_k^{\nu_{ki}''}](/images/math/0/8/b/08b53dce9957da556577c563ae8cc9d4.png)
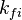 and
and 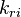 are the forward and reverse rate constant reaction i. Note that in OpenFoam the default value of the exponents in the above expression are the stoichiometric coefficients
are the forward and reverse rate constant reaction i. Note that in OpenFoam the default value of the exponents in the above expression are the stoichiometric coefficients ![[X]_k](/images/math/c/b/c/cbc7d65a80a572c296064e8d44295626.png) is the molar concentration of the species k. To continue with our example the reaction rates of the Zeldovich mechanism can be written:
is the molar concentration of the species k. To continue with our example the reaction rates of the Zeldovich mechanism can be written:
![\omega_1 = k_{f1}[NO][N] - k_{r1}[N2][O]](/images/math/a/7/0/a70bab94d73fb8ba6a8d65ebeb62aa68.png)
![\omega_2 = k_{f2}[N][O2] - k_{r2}[NO][O]](/images/math/2/2/c/22c6c68a7dd5ad6b945b412d1fe801a7.png)
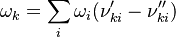
![\omega_k = \frac{d [X_k]}{dt} = \begin{pmatrix}\frac{d[N_2]}{dt} \\ \frac{d[O]}{dt} \\ \frac{d[NO]}{dt} \\ \frac{d[N]}{dt} \\ \frac{d[O_2]}{dt} \\\end{pmatrix} = \begin{pmatrix} -\omega_1 \\ -\omega_1 + \omega_2 \\ \omega_1 + \omega_2 \\ \omega_1 - \omega_2 \\ \omega_2 \\\end{pmatrix} = \begin{pmatrix} -(k_{f1}[NO][N] - k_{r1}[N2][O]) \\ -(k_{f1}[NO][N] - k_{r1}[N2][O]) + k_{f2}[N][O2] - k_{r2}[NO][O] \\ k_{f1}[NO][N] - k_{r1}[N2][O] + k_{f2}[N][O2] - k_{r2}[NO][O] \\ k_{f1}[NO][N] - k_{r1}[N2][O] - (k_{f2}[N][O2] - k_{r2}[NO][O]) \\ k_{f2}[N][O2] - k_{r2}[NO][O] \\\end{pmatrix}](/images/math/5/4/0/540092492d5d478241d0558e87b6766c.png)
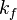 and
and 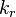 . The forward rate constant is calculated for Arrhenius type reactions as follows (see also
. The forward rate constant is calculated for Arrhenius type reactions as follows (see also
Cite error: <ref> tags exist, but no <references/> tag was found